Моногенные патологии — это такие заболевания, которые вызваны изменением только одного гена.
Ген представляет собой небольшой участок ДНК. Все гены находятся в хромосомах — длинных молекулах, расположенных внутри ядра клетки.
Ген представляет собой инструкцию, по которой организм формирует себя.
Генетические мутации могут возникать спонтанно или передаваться от родителей.
Виды наследования моногенных болезней

В медицине можно выделить различные типы наследования генетических патологий.
Основные виды наследования генетических заболеваний:
- Аутосомные — не зависят от пола ребенка;
- Сцепленные с полом ребенка.
Вероятность возникновения генетической мутации у потомства зависит от способа наследования болезни — является ли она аутосомно-доминантной или аутосомно-рецессивной.
При аутосомно-доминантном заболевании ребенок наследует от родителей одну нормальную копию гена и одну поврежденную. С вероятностью в 50% мутация будет «сильнее» и подавит здоровый ген.
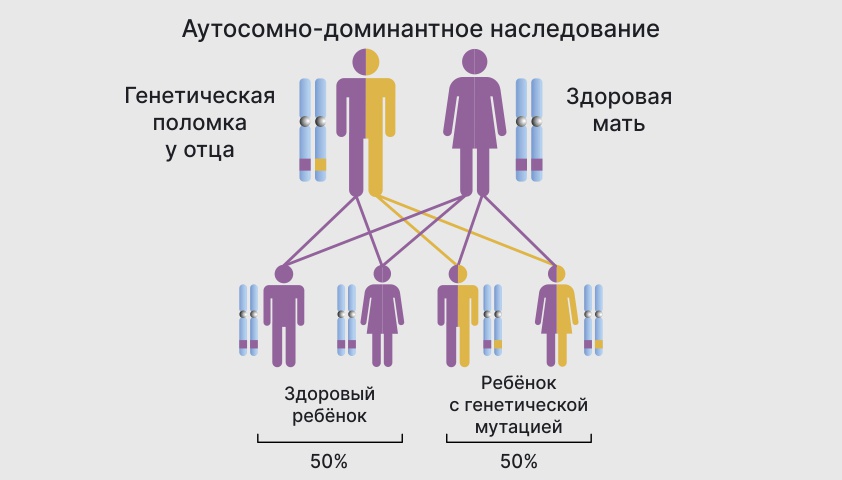
Вероятность рождения ребенка с патологией при аутосомно-доминантном наследовании — 50%
В случае аутосомно-рецессивного заболевания ребенок получает по копии поврежденного гена от каждого родителя — носителя мутации. У матери и отца признаков болезни может и не быть, но у ребенка заболевание проявится.
Шанс передачи рецессивного заболевания — 25%. В 50% случаев потомки с подобными мутациями становятся только переносчиками. В 25% случаев генетическое изменение не произойдет.
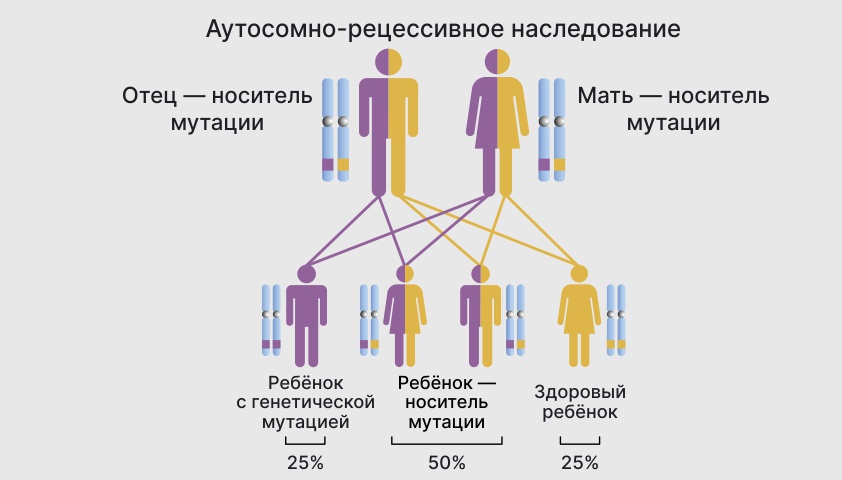
Вероятность появления ребенка с патологией при аутосомно-рецессивном типе наследования — 25%
Муковисцидоз и спинальная мышечная атрофия (СМА) входят в число наиболее распространенных аутосомно-рецессивных заболеваний.
Генетические болезни, сцепленные с полом, возникают, когда у ребенка происходит мутация в одной из половых хромосом — женской X или мужской Y.
Исследователи обнаружили, что один из генов X-хромосомы отвечает за цветовое восприятие. Если мальчик получает от матери копию такого гена с мутацией, то у него возникает дальтонизм: ребенок может различать лишь желтый и синий цвета, красный и зеленый кажутся ему одинаковыми.
Использование теста Ишихары поможет выявить наличие дальтонизма. Люди с таким нарушением зрения не смогут увидеть цифры 8, 9, 12, 7 на этом изображении
Носительство моногенных заболеваний
У носителей моногенных заболеваний одна копия гена повреждена. Вторая копия компенсирует дефект, и болезнь не проявляется.
Однако опасность носительства заключается в том, что человек может передать дефектный ген своим потомкам. В этом случае заболевание может проявиться у них.
Если оба родителя становятся носителями генетической мутации, то вероятность появления у них больного ребенка значительно возрастает.
Информация о моногенных заболеваниях
Частота моногенных заболеваний варьируется от 1 случая на 2 0000 новорожденных (муковисцидоз) до 1 случая на 350 0000 (анемия Фанкони).
Опасность таких заболеваний заключается в том, что ребенок может унаследовать их от внешне здоровых родителей — носителей патологии. При этом болезни протекают очень тяжело, а лекарства от них до сих пор не разработаны.
Часто встречающиеся моногенные заболевания
Известно около 5000 моногенных заболеваний, примерно половина из них наследуется по аутосомно-доминантному типу.
Наследственная нейросенсорная тугоухость — одно из самых распространенных моногенных заболеваний, оно обнаруживается у одного ребенка из 1000 новорожденных.
Причина заболевания — генетические изменения в следующих генах: SLC26A4, GJB2, LOXHD1, OTOGL, TMPRSS3, MYO6.
Заболевание обычно проявляется в первые месяцы жизни ребёнка. Основные признаки — ухудшение слуха и проблемы с речью.
Муковисцидоз (кистозный фиброз) — распространённое генетическое заболевание (1 случай на 2 000–2 500 новорождённых), вызванное мутациями в гене CFTR.
При муковисцидозе нарушается работа желез внешней секреции, таких как железы бронхолёгочной системы, поджелудочной железы, печени, потовых желез. Симптомы муковисцидоза связаны с тем, что секрет становится густым, вязким, из-за чего его выведение затрудняется. В результате могут возникнуть серьезные осложнения, такие как тяжелая пневмония, дыхательная и сердечная недостаточность, сахарный диабет, цирроз печени, лёгочные и желудочные кровотечения и другие проблемы.
Синдром Ушера типов 1C, 1D, 1B, 2A, 3A — уникальное заболевание, которое обнаруживается лишь у одного из 6 000 новорождённых. Причиной этого являются изменения в генах: USH2A, CLRN1, MYO7A, CDH23, USH1C, которые нарушают функционирование зрительных и слуховых рецепторов. В результате это приводит к постепенной потере слуха, ухудшению зрения и, в конечном итоге, к слепоглухоте.
Фенилкетонурия — редкое наследственное заболевание (возникающее лишь у 1 из 10 000 новорожденных), связанное с мутацией в гене PAH. Дети, страдающие от этого состояния, рождаются здоровыми. Однако они не способны усваивать фенилаланин — важную аминокислоту, содержащуюся в молоке, молочных смесях и прочих белковых продуктах. Со временем фенилаланин накапливается в теле и наносит вред нервной системе.
Первые признаки фенилкетонурии: тошнота, запах грибка от мочи и кожи, задержка развития умственных и двигательных способностей. Поздние симптомы: отставание в физическом развитии, судороги, экзема на коже.
Синдром Ретта — заболевание, которое диагностируется у одной из 10 000-15 000 новорожденных девочек (у мальчиков патология встречается крайне редко). Из-за изменений в гене MECP2 нарушается функционирование нервной системы, прогрессирует умственная отсталость, мышцы ослабевают. Дети с этим недугом не способны координировать свои движения, их позвоночник деформируется.
Врожденные нарушения гликозилирования типов 1A, 1B, 1C, 1K, 1P — группа генетически обусловленных заболеваний, вызванных мутациями в одном из генов: MPI, PMM2, ALG1, ATP7B, ALDOB. По статистике, такие патологии наблюдаются у одного ребенка из 10 000 новорожденных.
Гликозилирование — это процесс, в ходе которого к белкам организма при помощи ферментов присоединяются новые «строительные блоки» (остатки сахаров), образуя гликопротеины. Этот процесс является ключевым для нормального развития детей.
При нарушении процесса гликозилирования ребенок может столкнуться с задержкой в физическом и умственном развитии, нарушением свертываемости крови, а также появлением патологий сердца и печени.
Аутосомно-рецессивная поликистозная болезнь почек — унаследованное заболевание, диагностируемое у одного из 20 000 новорожденных. Причиной этого заболевания являются мутации в гене PKHD1.
Патология, вызванная генетическим дефектом, приводит к образованию множественных кист на поверхности и внутри почек. Эти кисты постепенно растут, замещая здоровые ткани, что приводит к потере способности почек нормально очищать кровь и выводить мочу. В итоге развивается хроническая почечная недостаточность.
Группа мукополисахаридозов 1H, 2, 3A, 3C представляет собой наследственные моногенные заболевания, вызванные мутациями в генах IDUA, IDS, HGSNAT, SGSH.
У пациентов с этими заболеваниями нарушается обмен веществ из-за недостатка определенных ферментов, необходимых для расщепления длинных молекул сахара — мукополисахаридов. Это приводит к накоплению сахара в тканях и органах, последующему их разрушению. Основные симптомы включают задержку физического развития, умственную отсталость, нарушения костей и хрящей, грыжи, увеличение печени и селезенки, а также сердечные проблемы.
Наследственная фруктозурия (непереносимость фруктозы) — достаточно редкое генетическое заболевание, встречающееся у одного ребёнка из 23 000–40 000. Из-за изменения в гене ALDOB нарушается метаболизм фруктозы — сахара, содержащегося, например, в мёде, яблоках, грушах, черносливе. В результате продукты её метаболизма накапливаются в организме и поражают многие органы и системы.

Людям с наследственной фруктозурией придётся исключить из питания продукты, содержащие фруктозу
Если человек с таким диагнозом употребит продукт, включающий в себя фруктозу, то у него возникнут тошнота, рвота и боль в животе. Без коррекции рациона, со временем разовьётся почечная и печёночная недостаточность.
Болезнь Баттена, также известная как нейрональный цероидный липофусциноз, связана с мутацией в одном из генов: TPP1, PPT1, CLN6. Частота появления этого заболевания составляет 1 случай на 25 000.
У пациентов, страдающих от болезни Баттена, в организме накапливается пигмент липофусцин, который токсично воздействует на клетки и разрушает их. Симптомы включают в себя приступы эпилепсии, ухудшение зрения, прогрессирующие двигательные и умственные нарушения.
Синдром Смита — Лемли — Опица — это нарушение обмена холестерина, вызванное дефектами в гене DHCR7. Это заболевание выявляется у одного из каждых 29 000 новорожденных.

Из-за генетического дефекта у ребенка могут возникнуть аномалии развития, например, дополнительные пальцы на руках (полидактилия)
Основные признаки данного наследственного заболевания: замедленный рост, умственная задержка, нарушение формирования пальцев на руках и ногах, а также дефекты развития внутренних органов.
Гепатоцеребральная дистрофия Вильсона — Вестфаля — Коновалова — редкое генетическое заболевание, вызванное изменениями в гене ATP7B. Частота встречаемости: 1 случай на 30 000–100 000 новорожденных.
У пациентов с данным заболеванием медь не выводится из организма, что приводит к её накоплению во внутренних органах и последующему разрушению. Люди с гепатоцеребральной дистрофией Вильсона — Вестфаля — Коновалова часто сталкиваются с проблемами сердца и печени, нарушениями свёртываемости крови и неврологическими расстройствами.
Галактоземия — это аномалия, которая возникает из-за изменений в гене GALT. Генетические дефекты приводят к тому, что организм не может усваивать галактозу — сахар, содержащийся в большинстве молочных продуктов.
У детей, страдающих от галактоземии, признаки проявляются уже с рождения — желтуха, увеличение печени, нистагм (ровные движения глаз), судороги, рвота. Со временем появляется катаракта. Ребенок замедляется в физическом и умственном развитии.
Частота встречаемости этого заболевания составляет 1 случай на 40 000–60 000 новорожденных.
Нейрофиброматоз II типа — это редкое (1:50 000) моногенное аутосомно-доминантное заболевание, вызванное изменениями в гене NF2.
У пациентов, страдающих от данного заболевания, происходит нарушение функционирования белка нейрофибромин-2, который обычно подавляет рост опухолей. Из-за неполадок в работе этого белка возникают несколько доброкачественных опухолей, которые возникают в центральной нервной системе и по ходу периферических нервов.
Тирозинемия I типа — это чрезвычайно редкое заболевание (1 случай на 100 000-120 000 новорожденных), которое развивается у людей с мутациями в гене FAN. У пациентов с таким диагнозом отсутствует фермент, участвующий в метаболизме аминокислоты тирозина. В результате в крови накапливаются токсичные вещества, которые негативно влияют на печень и нарушают свертываемость крови. У детей с тирозинемией часто наблюдается отставание в умственном и физическом развитии.
Болезнь, которая называется Андерсон — Фабри, это генетическое заболевание, вызванное нарушением GLA гена. Частота появления этой болезни составляет 1 случай на 117 000 новорожденных.
У людей с болезнью Андерсон — Фабри происходит недостаток фермента α-галактозидазы A. В органах скапливаются жирные продукты обмена веществ, которые в первую очередь начинают разрушать почки, сердце и сосуды головного мозга.
Болезнь Тейа — Сакса — очень редкое заболевание, которое обнаруживается всего у одного из 120 000 новорожденных (у евреев ашкенази частота гораздо выше — 1 случай на 3 000 человек).

Ашкенази — это этническая группа евреев, исторически населяющая Центральную и Восточную Европу
Из-за изменений в гене HEXA в нейронах (нервных клетках) накапливаются фрагменты клеточных оболочек, что приводит к постепенному разрушению нейронов, поражая головной и спинной мозг.
Основные симптомы болезни Тея-Сакса включают в себя затруднения с глотанием, нарушение фокусировки взгляда, нарушение двигательной активности, ухудшение зрения и слуха, отставание в умственном развитии, а также судороги.
Анемия Фанкони — генетическое заболевание, связанное с мутациями в одном из генов: FANCA, FANCC, FANG. Частота возникновения составляет 1 случай на 350 000 новорожденных.
При анемии Фанкони нарушается процесс образования крови, образуются злокачественные опухоли, развиваются врожденные дефекты. Симптомы заболевания включают частые кровотечения, синяки на коже, усталость, бледность, повышенная склонность к инфекциям.
Диагностика генетических заболеваний
Помощь в выявлении наличия мутаций в генах и оценке риска появления у ребенка моногенного заболевания оказывают генетические исследования.
Поиск изменений в одном из генов, вызывающих наследственную нейросенсорную тугоухость (SLC26A4, GJB2, LOXHD1, OTOGL, TMPRSS3, MYO6), и подтверждение или опровержение этого диагноза помогает целенаправленный анализ.
Диагностика муковисцидоза осуществляется специализированными генетическими исследованиями.
Выявление синдрома Ушера типов 1C, 1D, 1B, 2A, 3A — серьезного моногенного заболевания, которое чаще всего приводит к слепоглухоте у ребенка, — позволяет провести тест на генетические мутации.
Определение изменений в генах, способных вызвать фенилкетонурию, осуществляется с помощью генетического теста.
Выявление синдромов Ретта, синдрома Смита-Лемли-Опица, а также болезни Вильсона-Вестфаля-Коновалова, болезни Баттена, болезни Тея-Сакса и болезни Андерсона-Фабри помогает провести точные генетические исследования, которые доступны в Лаборатории Гемотест.
Генетические тесты также могут помочь выявить наследственную непереносимость фруктозы, чтобы разработать подходящий рацион вовремя.
Лаборатория Гемотест предоставляет возможность сдать анализы на самые редкие генетические патологии, включая аутосомно-рецессивную болезнь почек, мукополисахаридоз, нейрофиброматоз II типа и другие.
Для тех, кто планирует беременность или раздумывает стать донором спермы или яйцеклеток, целесообразно пройти комплексное исследование на предмет наличия наиболее распространенных генетических заболеваний.
Рекомендации для носителя после диагностики
Лишь одна копия гена имеет мутацию у носителя генетического заболевания. Такой индивидуум, как правило, находится в хорошем здоровье и не страдает от симптомов болезни, поскольку незаскомплектованный ген компенсирует дефект.
Согласно статистике, у большинства людей есть некоторое количество генетических заболеваний.
Однако если у партнеров, планирующих ребенка, гены с одинаковой мутацией, то вероятность появления болезни у младенца очень высока. Генетические исследования позволяют заранее оценить риск рождения больного ребенка во время планирования беременности.
Источники

- Бочков Н.П. Клиническая генетика. М., 2001.
- Томсон Г., Эспозито М.С. Генетика сложных заболеваний // Тенденции в клеточной биологии. 1999. Том 9(12). С. М17—20.
Видео по теме:
Вопрос-ответ:
Что такое моногенные заболевания?
Моногенные заболевания — это группа заболеваний, вызванных изменениями в одном гене. Такие изменения передаются от родителей к потомкам и могут привести к различным патологиям и синдромам.
Какие примеры моногенных заболеваний существуют?
Примерами моногенных заболеваний являются цистический фиброз, муковисцидоз, ДЦП (дистрофия сетчатки), наследственные формы рака, болезни Паркинсона и Хантингтона. Всего существует более 6000 моногенных заболеваний.
Каковы основные причины возникновения моногенных заболеваний?
Основными причинами возникновения моногенных заболеваний являются мутации в генах, ответственных за кодирование определенных белков. Такие мутации могут возникнуть случайно или быть унаследованы от родителей. В данном случае гены не могут исправлять ошибки, что приводит к возникновению заболевания.
Какие методы диагностики используются для выявления моногенных заболеваний?
Для диагностики моногенных заболеваний могут использоваться методы молекулярной генетики, такие как секвенирование ДНК, ПЦР, ФЛГ или микрочип-гибридизация. Также может применяться генетическое тестирование и анализ патогенных мутаций.
Каковы перспективы лечения моногенных заболеваний?
Лечение моногенных заболеваний может быть направлено на коррекцию генетического дефекта, замену поврежденного гена или терапию заменой белка. Также исследуются методы генной терапии и терапии клеточными технологиями. Однако эффективные методы лечения пока еще находятся на стадии разработки и испытаний.
Что такое моногенные заболевания?
Моногенные заболевания — это группа генетических заболеваний, которые вызваны дефектом или мутацией в одном гене. Эти заболевания наследуются по принципу моногенного наследования, то есть передаются от родителей к потомкам по определенной схеме.